A Novel Computational Strategy for miRNA Gene – Disease Relationship Prediction Using Locality-Constrained Linear Coding

MiRNAs are small non-coding regulatory RNAs that are related to a number of ailments. Rising proof has proven that miRNAs play necessary roles in numerous organic and physiological processes. Subsequently, the identification of potential miRNA-disease associations might present new clues to understanding the mechanism of pathogenesis. Though many conventional strategies have been efficiently utilized to find a part of the associations, they’re normally time-consuming and costly. Consequently, computational-based strategies are urgently wanted to foretell the potential miRNA-disease associations in a extra environment friendly and resources-saving method. On this paper, we suggest a novel methodology to foretell miRNA-disease associations based mostly on Locality-constrained Linear Coding (LLC).
Why miRNA-Disease Prediction Matters
miRNAs regulate gene expression by binding to messenger RNA (mRNA) targets, silencing gene expression or inhibiting translation. Aberrations in miRNA expression profiles are linked to a spectrum of diseases, positioning them as biomarkers for early diagnosis, prognosis, and therapeutic targets.
However, experimental validation of miRNA-disease associations is time-consuming and costly. Hence, computational prediction models have become indispensable, enabling the discovery of potential associations prior to wet-lab validation.

Community-based affiliation evaluation to deduce new illness–gene relationships utilizing large-scale protein interactions.

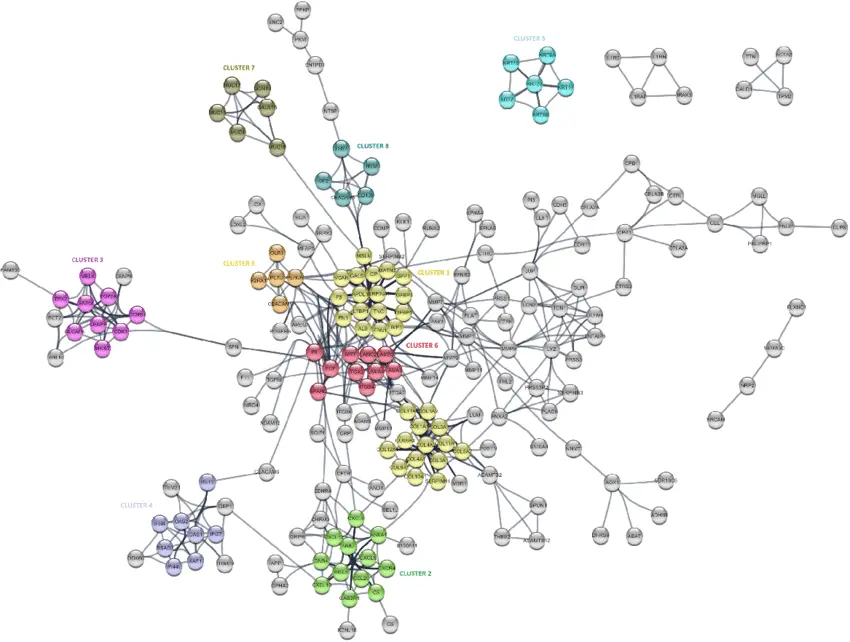
Protein-protein interactions built-in with disease-gene associations symbolize necessary data for revealing protein features beneath illness circumstances to enhance the prevention, prognosis, and remedy of advanced ailments. Though a number of research have tried to determine disease-gene associations, the variety of doable disease-gene associations may be very small.
Excessive-throughput applied sciences have been established experimentally to determine the affiliation between genes and ailments. Nonetheless, these strategies are nonetheless fairly costly, time consuming, and even troublesome to carry out. Thus, based mostly on at the moment obtainable knowledge and information, computational strategies have served as alternate options to offer extra doable associations to extend our understanding of illness mechanisms. Right here, a brand new network-based algorithm, particularly, Illness-Gene Affiliation (DGA), was developed to calculate the affiliation rating of a question gene to a brand new doable set of ailments.
First, a large-scale protein interplay community was constructed, and the connection between two interacting proteins was calculated with regard to the illness relationship. Novel believable disease-gene pairs had been recognized and statistically scored by our algorithm utilizing neighboring protein data. The outcomes yielded excessive efficiency for disease-gene prediction, with an F-measure of 0.78 and an AUC of 0.86. To determine promising candidates of disease-gene associations, the affiliation protection of genes and ailments had been calculated and used with the affiliation rating to carry out gene and illness choice. Primarily based on gene choice, we recognized promising pairs that exhibited proof associated to a number of necessary ailments, e.g., irritation, lipid metabolism, inborn errors, xanthomatosis, cerebellar ataxia, cognitive deterioration, malignant neoplasms of the pores and skin and malignant tumors of the cervix.
Specializing in illness choice, we recognized goal genes that had been necessary to blistering pores and skin ailments and muscular dystrophy. In abstract, our developed algorithm is straightforward, effectively identifies disease-gene associations within the protein-protein interplay community and gives further information relating to disease-gene associations. This methodology might be generalized to different affiliation research to additional advance biomedical science.
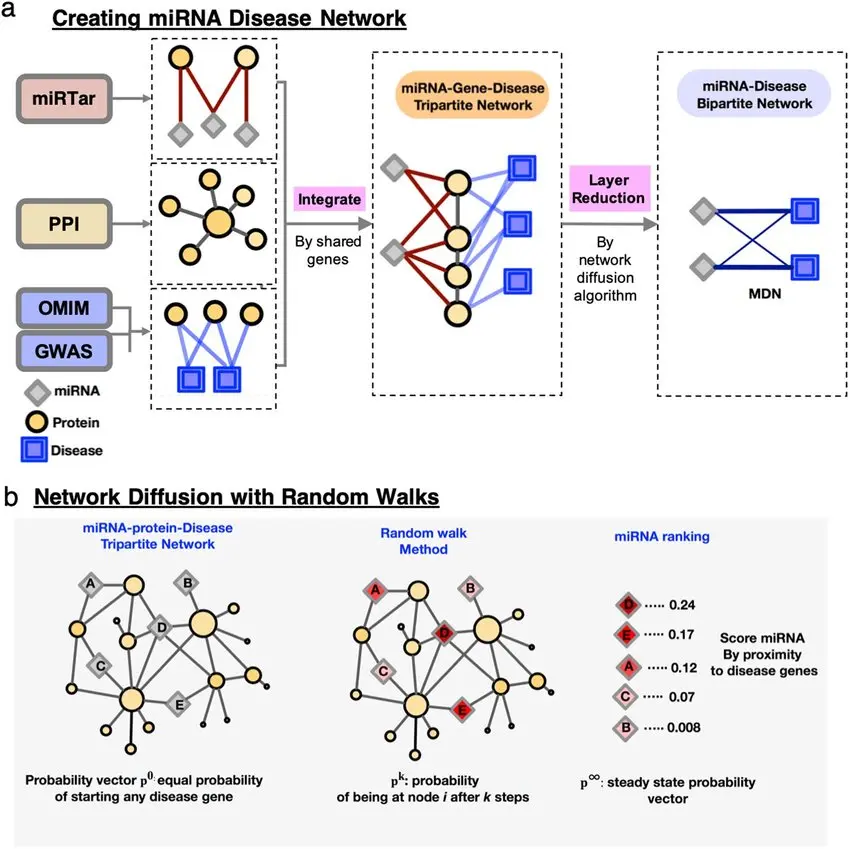
Relationship between Deleterious Variation, Genomic Autozygosity, and Illness Danger: Insights from The 1000 Genomes Undertaking.
Regions of autozygosity (ROAs) are genomic segments that are homozygous due to identical-by-descent (IBD) inheritance from a common ancestor. These regions are unevenly distributed across the genome and have been associated with increased risk for several complex diseases. This study builds on earlier findings by analyzing whole-genome sequencing data from 2,436 individuals in the 1000 Genomes Project, confirming that long ROAs are significantly enriched for deleterious homozygous variants especially strongly damaging ones due to recent parental relatedness and limited purifying selection. The enrichment levels vary widely across populations and are particularly pronounced in genes associated with Mendelian diseases and FDA-approved drug targets. Moreover, genes implicated in various complex traits also exhibit elevated levels of damaging homozygotes in ROAs, although the patterns differ by disease and population. These findings underscore the importance of considering population genetic background when interpreting associations between ROAs and complex disease risks, as it may explain inconsistencies in previous ROA-disease association studies.
Current Computational Landscape: Limitations
Many traditional computational models such as similarity-based methods, matrix factorization, network propagation, or machine learning classifiers have improved prediction accuracy. Yet, they often struggle with
High-dimensional data
Overfitting and generalization issues
Non-linear relationships
Data sparsity

What Is Locality-Constrained Linear Coding (LLC)?
Originally used in image recognition, LLC is a feature encoding technique that emphasizes locality over global sparsity. It projects high-dimensional data points into a lower-dimensional subspace while preserving the local geometric structure of the data.
In simpler terms, LLC encodes each data point as a linear combination of its nearest neighbors, rather than the entire dataset. This makes it especially powerful for:
Capturing localized patterns
Reducing noise and redundancy
Enhancing model interpretability and generalization

Proposed Method: LLC for miRNA-Disease Prediction
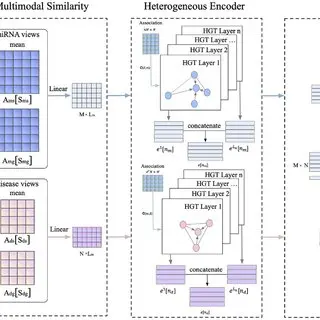
Data Integration and Feature Construction
The model integrates diverse biological data:
miRNA functional similarity networks
Disease semantic similarity networks
Known miRNA-disease associations
Gene expression and sequence information (optional enhancement)
Locality-Based Codebook Generation
Using a k-nearest neighbors (k-NN) strategy, a codebook of local bases is constructed. Each miRNA (or disease) is then expressed as a linear combination of its local neighbors.
This step retains the local manifold structure, ensuring that similar miRNAs or diseases are encoded similarly, even if the global structure is non-linear.
LLC Encoding and Optimization
LLC performs linear coding under locality constraints by solving:
minC∑i∥xi−Bci∥2+λ∥di⊙ci∥2s.t.1Tci=1\min_{C} \sum_{i} \left\| x_i - B c_i \right\|^2 + \lambda \left\| d_i \odot c_i \right\|^2 \quad \text{s.t.} \quad \mathbf{1}^T c_i = 1Cmini∑∥xi−Bci∥2+λ∥di⊙ci∥2s.t.1Tci=1
Where:
xix_ixi = feature vector of sample i
BBB = local codebook bases
cic_ici = encoding coefficients
did_idi = locality adaptor (higher for closer neighbors)
λ\lambdaλ = regularization term
Prediction and Ranking
Once encoded, association scores between miRNAs and diseases are computed using:
Cosine similarity or distance-based metrics
Optional: supervised classifiers (e.g., SVMs, logistic regression) trained on the LLC-transformed features